Current mainline workflow
Data and feature construction
The user-provided molecule-solvent table is cleaned and converted into Morgan fingerprints, RDKit fragment-count features, and solvent one-hot features.
Training-set feature screening
The data are split into training and held-out test subsets. Feature filtering and recursive feature elimination are performed only on the training set, producing the fixed feature table used by the retrained model.
Candidate model comparison
Eight regressors are compared by 10-fold cross-validation on the training set: CatBoost, XGBoost, LightGBM, RF, GBR, KNN, KRR, and SVR. The best-performing model can then be selected as the final base model; the template uses XGBoost.
Final prediction and residual correction
The default workflow trains a tuned XGBoost base model. If users add an OOF residual library, the web app applies a gated nearest-neighbor correction using the parameters supplied in NN_Residual_Correction_Config.json.
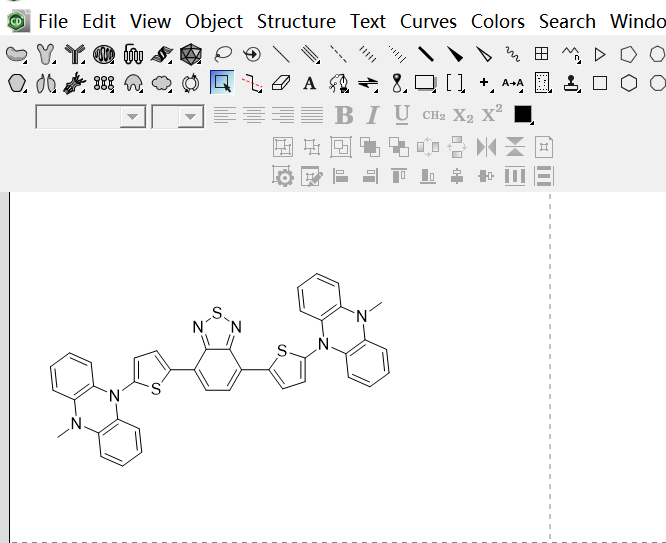
Draw the molecule structure in ChemDraw.
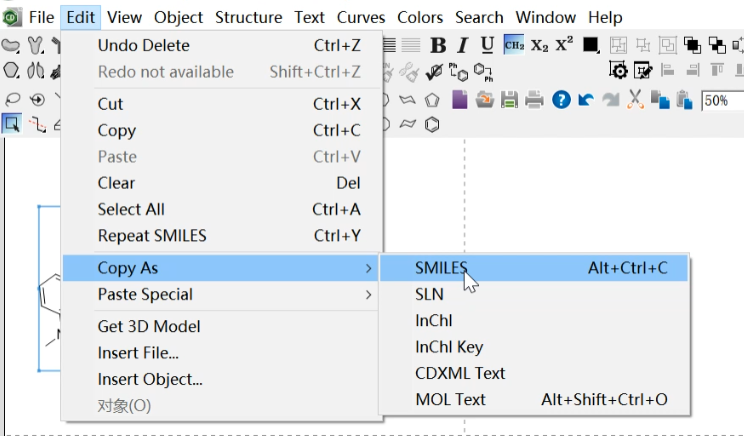
Select the molecule and copy it as SMILES.

Input or choose a solvent label from common solvents.

After prediction, the emission wavelength is shown in nm.
Solvent abbreviations
Please note that only the following solvents use abbreviations:
| Abbreviation | Full Name |
|---|---|
| THF | Tetrahydrofuran |
| DCM | Dichloromethane |
| TOL (PHME) | Toluene |
| H2O | Water |
| DMSO | Dimethyl sulfoxide |
| MEOH | Methanol |
| ACN | Acetonitrile |
| CFM (TCM) | Chloroform |
| HEX | n-Hexane |
| ETOH | Ethanol |
| VAC | Vinyl acetate |
| ACOET | Ethyl acetate |
| CHX | Cyclohexane |
| DMF | N,N-Dimethylformamide |
| DIOX | 1,4-Dioxane |
| AC | Acetone |
| IPROPOH (IPA) | Isopropanol |
| CBZN | Chlorobenzene |
| BZN | Benzene |
| SOLID | Solid state |
| THF/H2O | THF-Water mixture |
| PBS | Phosphate buffered saline |
| BZNIT | Benzonitrile |
| DCE | 1,2-Dichloroethane |
| BLUME | Butyl methyl ether |
| DEE | Diethyl ether |
| MXYLENE | m-Xylene |
Why are predicted results different from experimental results?
The discrepancy between the predicted and experimental results can arise from two main aspects. First, from the data perspective, the molecule under investigation may contain novel structural motifs that are underrepresented or absent in the training set of BTD-EmisPred. Second, from the model perspective, all empirical models have intrinsic prediction errors. Our model learns statistical correlations from experimental data, which inevitably contain measurement uncertainties. Therefore, predicting a perfect match for a new query molecule is challenging. We are actively expanding the training database and exploring strategies to quantify prediction confidence to better address these limitations.
Please let us know your molecule, solvent, and optical properties by sending an email to gaowen@sdnu.edu.cn. We will add your molecules to our database as soon as possible.